High-Throughput Lead Optimization with Amber
Project Description
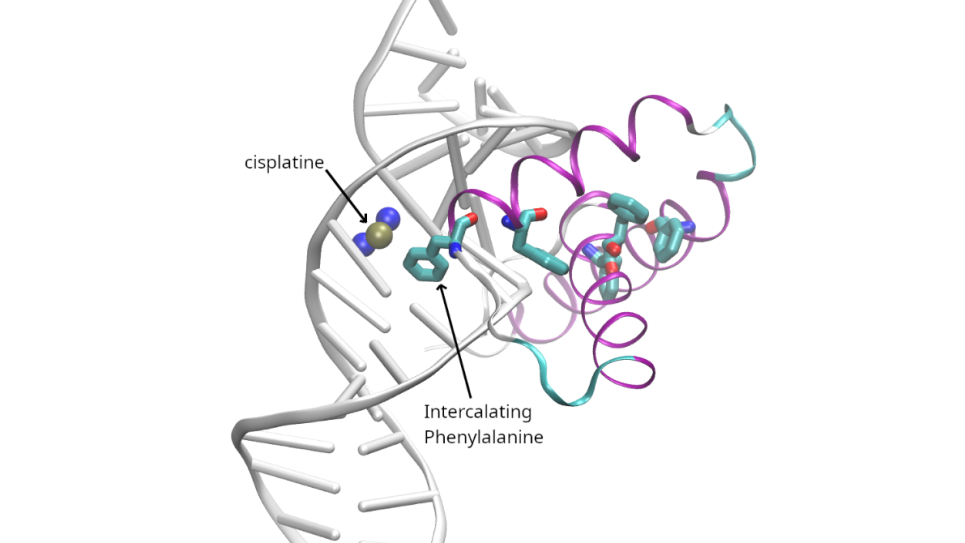
The overall objective of this project is to address a major obstacle in computer-aided drug discovery and precision medicine: the absence of computational workflows capable of reliably identifying the most tight-binding ligands from a design pool of proposed compounds within 60 hours of wall-clock time (a typical time range required for a drug discovery team in industry). The researchers will develop and test a new adaptive framework for high-throughput lead optimization that integrates advanced methods and features in AMBER to achieve ligand-protein binding predictions. The framework will be optimized to run with unprecedented efficiency on advanced GPU supercomputing platforms such as Polaris.