Materials — we use them, wear them, eat them and create them. Sometimes we invent them by accident, like with Silly Putty. But far more often, making useful materials is a tedious and expensive process of trial and error.
Scientists at the U.S. Department of Energy’s (DOE) Argonne National Laboratory have recently demonstrated an automated process for identifying and exploring promising new materials by combining machine learning (ML) — a type of artificial intelligence — and high performance computing. The new approach could help accelerate the discovery and design of useful materials.
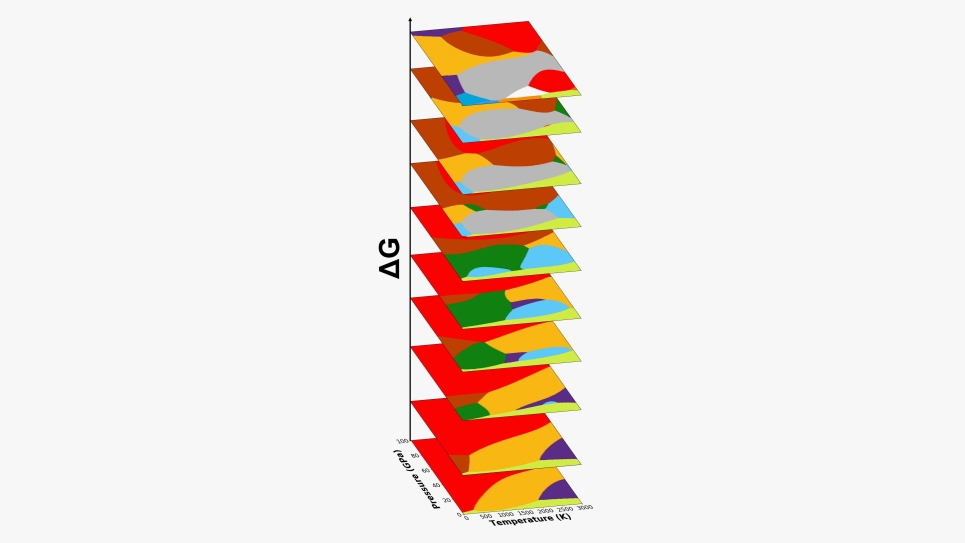
Using the single element carbon as a prototype, the algorithm predicted the ways in which atoms order themselves under a wide range of temperatures and pressures to make up different substances. From there, it constructed a series of what scientists call phase diagrams — a kind of map that helps guide their search for new and useful states of matter.
“We trained a computer to probe, question and learn how carbon atoms could be organized to create phases that we might not find on earth or that we don’t fully understand, thereby automating a whole step in the materials development process,” said Pierre Darancet, an Argonne scientist and author on the study. “The more of this process a computer can handle on its own, the more materials science we can get done.”
Equilibrium and beyond
When a material’s atomic structure changes, so can its electronic, thermal and mechanical properties. Scientists want to find new ways to arrange atoms to develop useful materials. A prime way to change the atomic structure of a material is to vary the surrounding pressure and temperature.
We see this sort of structural change commonly in water. At room temperature and normal atmospheric pressure, water is most stable as a liquid. If you decrease the temperature enough, the same water molecules will arrange themselves to form solid ice.
Similarly, diamond and graphite are wildly different materials, but they both consist exclusively of carbon atoms — just arranged in different ways. Under normal conditions, graphite is a much more stable form of carbon than diamond. Under conditions of extreme pressure and heat, however, graphite slowly crystallizes into diamond. When removed from those extreme conditions, the diamond persists, lingering in what scientists call a metastable state.
The ML algorithm constructed phase diagrams that mapped hundreds of these metastable states of carbon, some known and some new.
“It’s experimentally difficult to predict and produce states of matter that are not near equilibrium conditions,” said Jianguo Wen, an Argonne experimentalist on the study. “This new computational approach enables us to explore those little-known regions on the maps that aren’t otherwise accessible, or that we don’t yet even know exist.”
Verifying the algorithm
The scientists trained the ML algorithm with synthetic data, which is produced through simulation and approximates results scientists would get through experiment. They generated the dataset using molecular dynamics and density functional theory, both common computational chemistry tools.
The training data was produced using Carbon, a high performance computing cluster at Argonne’s Center for Nanoscale Materials (CNM), a DOE Office of Science user facility. Also used were two other DOE user facilities: the Argonne Leadership Computing Facility and the National Energy Research Scientific Computing Center at Lawrence Berkeley National Laboratory.
Using the algorithm’s predictions as a guide, the team verified its efficacy by synthesizing actual samples and characterizing them using a transmission electron microscope at CNM.
The algorithm successfully predicted well-known phase diagrams for carbon, and the computer-generated phase diagrams affirmed and shed light on several yet-unexplained experimental observations.
In particular, the algorithm identified the previously ambiguous structure of n-diamond (stands for “new diamond”), a state of carbon that has mystified scientists since it was theorized over 30 years ago. “The algorithm made new and surprising predictions about structural features of n-diamond that we verified with experiment, demonstrating that the algorithm holds up, even with high-profile phases,” said Wen.
The team also synthesized several phases predicted by the algorithm that have not yet been reported in the scientific literature. The structures of the samples matched the predictions, further verifying the algorithm.
“Materials synthesis, especially of those with exotic properties, can often take several experimental trials and years of effort,” said Argonne scientist Subramanian Sankaranarayanan, a lead author on the study. “Our machine learning algorithms allow us to identify the synthesis conditions of exotic materials, potentially reducing the time for their experimental realization.”
In this study, the algorithm was applied to carbon only. In the future, the scientists hope to apply the same approach to systems of more than one element. Applying the machine learning algorithm to more complex systems could lead to broad impact on the discovery and design of useful materials.
A paper on the study, “Machine learning the metastable phase diagram of covalently bonded carbon,” was published in Nature Communications. In addition to Darancet, Sankaranarayanan and Wen, Argonne authors include Rohit Batra, Henry Chan, Troy Loeffler, Duan Luo, Sukriti Manna and Srilok Srinivasan.
This work was supported by the DOE Office of Basic Energy Sciences, Data, Artificial Intelligence and Machine Learning through the DOE Scientific User Facilities program (Digital Twins and Collaborative MLExchange). Additional computing resources included Fusion and Blues, high performance computing clusters operated by the Laboratory Computing Resource Center at Argonne.
==========
About Argonne’s Center for Nanoscale Materials
The Center for Nanoscale Materials is one of the five DOE Nanoscale Science Research Centers, premier national user facilities for interdisciplinary research at the nanoscale supported by the DOE Office of Science. Together the NSRCs comprise a suite of complementary facilities that provide researchers with state-of-the-art capabilities to fabricate, process, characterize and model nanoscale materials, and constitute the largest infrastructure investment of the National Nanotechnology Initiative. The NSRCs are located at DOE’s Argonne, Brookhaven, Lawrence Berkeley, Oak Ridge, Sandia and Los Alamos National Laboratories. For more information about the DOE NSRCs, please visit https://science.osti.gov/User-Facilities/User-Facilities-at-a-Glance.
The Argonne Leadership Computing Facility provides supercomputing capabilities to the scientific and engineering community to advance fundamental discovery and understanding in a broad range of disciplines. Supported by the U.S. Department of Energy’s (DOE’s) Office of Science, Advanced Scientific Computing Research (ASCR) program, the ALCF is one of two DOE Leadership Computing Facilities in the nation dedicated to open science.
Argonne National Laboratory seeks solutions to pressing national problems in science and technology. The nation's first national laboratory, Argonne conducts leading-edge basic and applied scientific research in virtually every scientific discipline. Argonne researchers work closely with researchers from hundreds of companies, universities, and federal, state and municipal agencies to help them solve their specific problems, advance America's scientific leadership and prepare the nation for a better future. With employees from more than 60 nations, Argonne is managed by UChicago Argonne, LLC for the U.S. Department of Energy's Office of Science.
The U.S. Department of Energy's Office of Science is the single largest supporter of basic research in the physical sciences in the United States and is working to address some of the most pressing challenges of our time. For more information, visit https://energy.gov/science