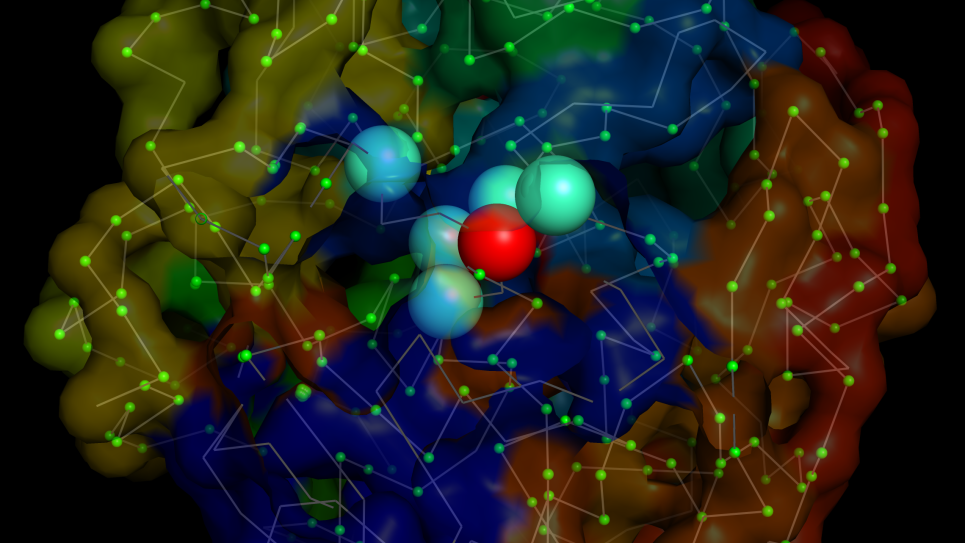
Single-cell 3D chromatin structure, reconstructed from population Hi-C data using polymer modeling, reveals complex, many-body interactions between a gene (red) and multiple regulatory elements (cyan). Image: Hammad Farooq and Jie Liang, University of Illinois Chicago.


